PERIPHERIN-2 (PRPH2) associated retinal degeneration

We are proud to be part of an exciting collaboration between the University of California San Diego and The Nixon Visions Foundation. This collaboration focuses on increasing the understanding and the development of breakthrough novel treatments for PRPH2 associated retinal degeneration (PARD).
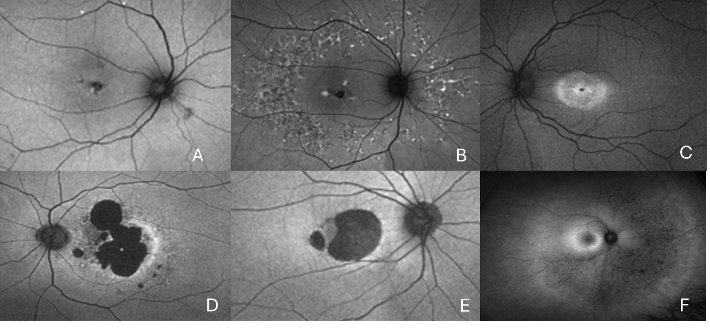
Retinal disease caused by mutations in the gene PRPH2 is one of the commonest causes of inherited retinal disease. PARD is complex as it causes a wide spectrum of different retinal diseases. However, all these diseases are progressive and can lead to severe visual impairment (Seff Figure 1).
PARD is usually inherited in an autosomal dominant manner. This means that there is a 1 in 2 chance of affected individuals passing the disease on to children. As a result, these sight-threatening diseases can afflict multiple generations of families.
At present there are no treatments for PRPH2-associated retinal degeneration (PARD). The understanding of PARD and the development of new treatments has been hampered by the relative lack of models which replicate clinical disease and so help clinical translation of treatments. This highlights a critical need to develop new clinically relevant models to better understand human disease and to leverage these insights to develop novel sight saving treatments.
The Borooah lab specializes in modelling inherited retinal disease and testing gene modifying treatments. We have previously developed, characterized and treated both animal and humanized retinal disease models.
As part of this collaboration our scientists first generated pluripotent stem cells from patients’ blood samples. Pluripotent stem cells allow the growing of almost all tissues within the body with the right guidance (See Figure 2).

Our scientists then differentiated these cells using our in-house protocols to generate ‘mini-eyes’, known as retinal organoids. (See Figure 3)
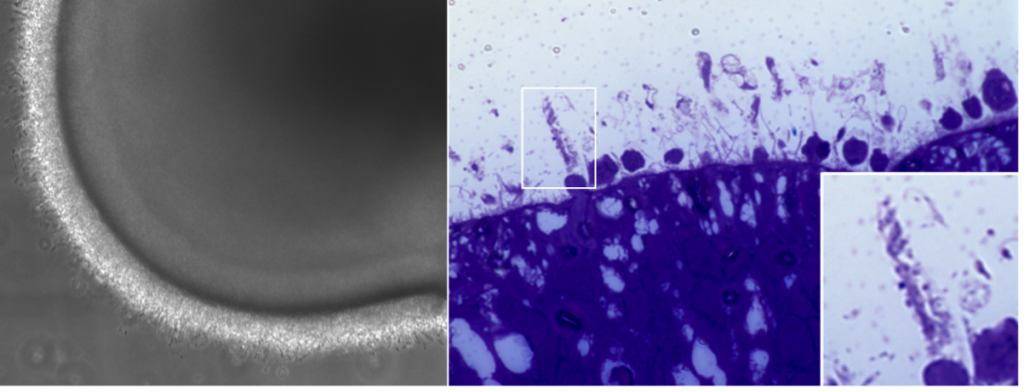
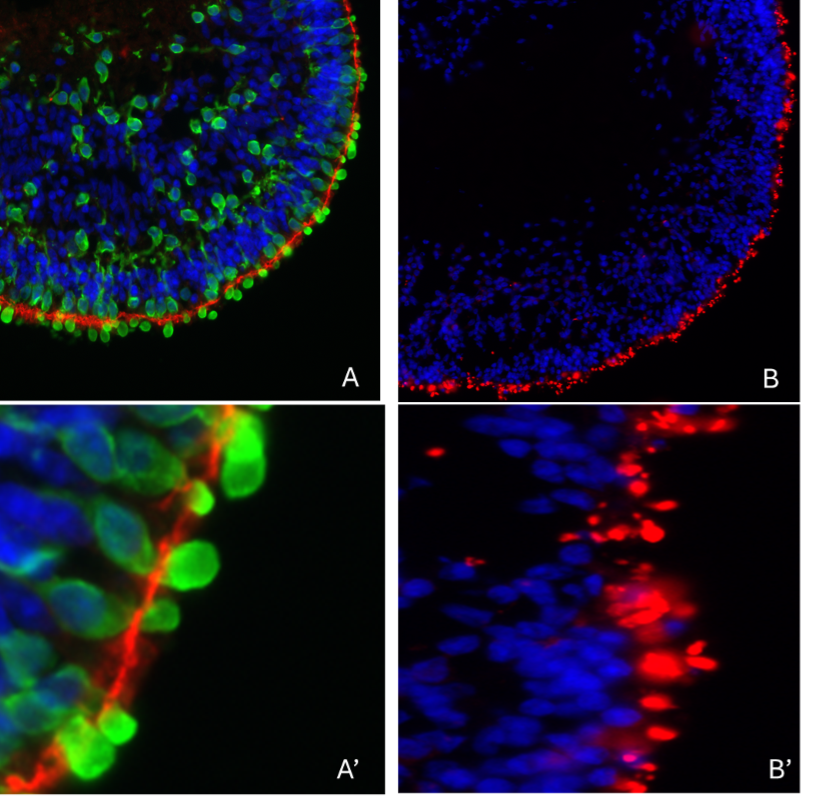
Retinal organoids develop most of the cells found in the adult human retina. Importantly, this includes photoreceptors, the main cells affected in PARD and so this is a good platform with which to study this disease. PRPH2 protein is mainly found in a specialized region known as the outer segment, in photoreceptors (See Figure 5).
The initial studies will initially focus on one particular mutation in PRPH2. This mutation mainly results in disease affecting the macula, the central part of the retina and the region of the retina important for central vision.
The studies have three main objectives:
Objective 1:
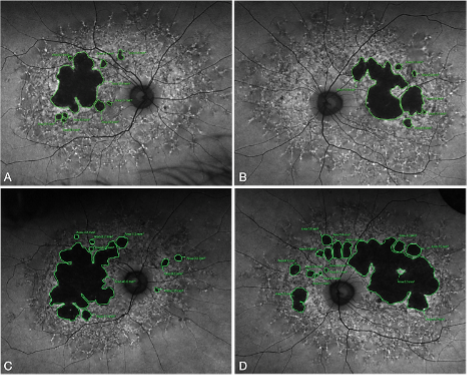
To understand the clinical effect of the specific mutation in patients with PRPH2 (See Figure 4)
Objective 2:
To understand the cone dominant/macular phenotype of this mutation and to investigate the causes of phenotypic variability using human retinal organoids.
Objective 3:
To test novel treatments using this organoid model

This collaboration is likely to benefit not only those with the mutation of interest but will also likely help the understanding of disease in general. It is expected that the studies will provide clinical and basic science tools to help clinical translation and ultimately assist the development of new treatments to prevent blindness from PARD.