Research
The focus of our research is to develop novel treatments for currently untreatable sight-threatening retinal and macular degenerations. Our research harnesses our strengths in both clinical imaging and basic laboratory research. By using multi-modal imaging and state of the art image analysis at the Jacobs retina center we phenotyped patients with different forms of retinal degeneration. In the laboratory, we model these patient diseases using our specialist expertise with eye stem cell and animal models to better understand the cause of disease.
Clinical Imaging & Analysis
Age-related Macular Degeneration
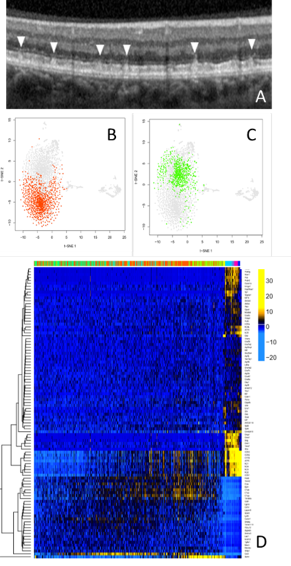
Age-related macular degeneration (AMD) is one of the most common causes of blindness in the developed world. Although treatments have been developed for ‘wet’ AMD, which is caused by bleeding at the back of the eye, there are currently no treatments for the most common form of AMD, ‘dry’ AMD. Dry AMD results from a slow loss of cells at the macula, the part of the retina responsible for central vision.
A common feature of dry AMD is a subretinal deposit called pseudodrusen. Patients with pseudodrusen suffer from an increased rate of macular degeneration. Little is known about why pseudodrusen develop and how they exacerbate macular degeneration. The study of pseudodrusen and the development of treatments has been limited by a lack of suitable models.
Recently we studied an animal that developed deposits similar to pseudodrusen. We identified abnormally activated immune cells to play a role in the early formation of these subretinal deposits. Similar immune cell findings have also recently been reported in AMD patients. To better understand the cause of this subretinal immune cell activation we developed a technique to study individual retinal immune cells. These studies identified the cascade of gene changes in individual cells which may cause the immune cell activation associated with retinal degeneration in this model.
The next aim is to repurpose FDA approved immunomodulatory drugs that specifically target the ‘bad’ immune cells identified in our studies while sparing the ‘good’ immune cells responsible for immune protection in our model and if successful will aim to translate successful treatments to human clinical trial to treat patients in early AMD with pseuoddrusen in order to slow macular degeneration.
Early-onset Inherited Retinal Degenerations
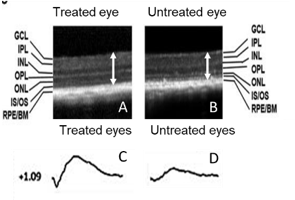
(A,B) Retinal scans in the mouse model show that there is a clear difference in retinal thickness in the treated eye (A). compared to the untreated eye (B). (C,D) Similarly, electroretinography studies in the treated eye show a greater electrical response (c) compared with the untreated eye (D)
Inherited retinal disease is a major cause of untreatable childhood blindness. We inherit a set of genes from each of our parents. There are now over 300 genes associated with retinal disease. Most early-onset retinal degenerations result from recessive disease. Recessive disease occurs when we lose both sets of our inherited genes due to gene defects known as mutations.
One way of treating recessive disease is by gene replacement therapy. Gene replacement therapy provides the missing genes allowing cells to function normally. This approach has recently been successfully used for the first FDA approved treatment for inherited retinal disease in another early-onset retinal degeneration,called Leber Congential Amarousis.
We have recently described a new model of early-onset retinitis pigmentosa caused by mutations in the gene mouse frizzled receptor protein (MFRP). The model replicates many of the features of human disease including early-onset retinal degeneration and a loss of retinal electrical response. We recently tested gene therapy to treat this model. We found that treatment using gene therapy was able to provide long-term rescue of both retinal degeneration and retinal electrical function in the model. Successful long-term rescue of vision in the animal model is an important translational step. Using a similar approach we plan to pursue clinical trials to treat patients with the similar genetic diseases to attempt to rescue their remaining vision. This work is in collaboration with Dr. Radha Ayyagari.
Late-onset Inherited Retinal Degenerations
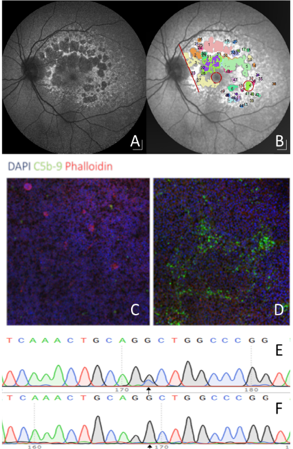
Late-onset inherited retinal disease is a cause of severe visual loss in the older population. Late-onset disease results from a slower-onset retinal degeneration and is more commonly linked to dominant inherited disease. Dominant diseases are usually caused by a single faulty gene which disrupts normal retinal function.
One approach to treat a dominant disease is to use gene editing. By targeting the faulty gene using CRISPR-Cas9 gene editing the faulty gene can be removed allowing the retina to function properly with the remaining normally functioning gene. We have recently developed a model of retinal degeneration caused by the a disease called late-onset retinal degeneration (L-ORD). This dominant disease causes blindness in patients in their 60s and shares many similarities with AMD. We have recently completed a long-term natural history study to describe the progression of disease in eyes of patients to identify the best time to treat patients. This work is in close collaboration with Professor Baljean Dhillon, Professor Siddharthan chandran and Dr Andrew Browning.
Recently, we also developed a lab model of L-ORD using patient stem cells. We found that patient cells, carrying the mutations, developed a marked immune activation similar to that seen in patient retinal pathology samples. We then edited the mutation using CRISPR-Cas9 and found that the cells normalized their immune activation, providing a powerful in vitro phenotype. We have recently developed a virus based approach to treat our stem cell model and an animal model. If successful, the plan is to translate this strategy to treat patients leveraging our well phenotyped cohort of patients to treat patients prior to sight loss.
Summary
The laboratory is using a variety of retinal specific tools and techniques to make progress in the prevention and treatment of vision loss. This bench side work will be closely aligned with work in the new retinal degeneration clinic which will characterize patient disease using the latest multi-modal imaging and genetic testing with the aim of translating novel therapies to preserve vision.